Diagnosis and management of neurogenic orthostatic hypotension
Article information

Abstract
Orthostatic hypotension is a sustained and pathological drop in blood pressure upon standing. Orthostatic hypotension can be due to non-neurogenic conditions or autonomic disorders. Impaired baroreflex-mediated vasoconstriction and insufficient release of norepinephrine play key roles in the pathophysiology of neurogenic orthostatic hypotension. Its common symptoms mainly related to inadequate cerebral blood flow include dizziness, lightheadedness, and syncope. It is crucial to differentiate neurogenic orthostatic hypotension from non-neurogenic orthostatic hypotension. For the management of neurogenic orthostatic hypotension, physicians should implement non-pharmacological methods and, if possible, reverse combined non-neurological conditions. Depending on severity of symptoms, pharmacological intervention may be tried after or with non-pharmacological methods. Its management should be individualized based on intensity of symptoms, comorbid conditions, drug side effects, and etiology. In this review, we discuss the definition, pathophysiology, clinical approach, and management of neurogenic orthostatic hypotension.
INTRODUCTION
Immediately after standing up, gravity moves 450-500 mL of blood downward in the intravascular space. With continued standing, there is a gradual shift of fluid from the intravascular to the interstitial space. Therefore, orthostatic stress (standing) causes a large fluid shift of approximately 500-1,000 mL, which results in increased blood pooling in the dependent parts of the body below the heart. The main areas of blood pooling are the intestine and the large muscle groups of the legs.1-4 To keep adequate blood supply to the brain, the autonomic nervous system plays a key role by activating various compensatory mechanisms, such as vasoconstriction of blood vessels in the non-essential areas. The arterial baroreflex is the main mechanism for autonomic blood pressure (BP) control. Mechanoreceptors in the carotid artery monitor changes in the stretch of the arterial walls and send afferent signals to the brainstem. After processing the signals, the brainstem sends out efferent signals to the main target organs changing heart rate (HR) and blood vessel tone accordingly.5,6 Furthermore, prolonged upright posture stimulates the renin-angiotensin-aldosterone system and vasopressin secretion regulating blood volume on a long-term basis and ultimately maintaining cerebral blood flow.6,7 Failure of these compensatory mechanisms can lead to orthostatic hypotension (OH). Inadequate cerebral perfusion causes dizziness, lightheadedness, vision changes and syncope/pre-syncope. In this review article, we will discuss abnormal hemodynamic responses to standing up including OH, its pathophysiology, symptoms and management.
ABNORMAL HEMODYNAMIC CHANGES UPON STANDING UP
There exist four types of abnormal drops in BP after standing up: classic OH, initial OH, delayed OH, and delayed BP recovery.3,8 Exaggerated postural tachycardia or postural orthostatic tachycardia syndrome is not discussed here because there is no pathological drop in BP by its definition.8
Classic orthostatic hypotension
Classic OH is defined as a sustained reduction of systolic blood pressure (SBP) by at least 20 mmHg or of diastolic blood pressure (DBP) by at least 10 mmHg within 3 minutes of standing or tilt table testing with at least 60 degrees of head-up tilt (Fig. 1A, B).1-3,6-8 In hypertensive patients (a supine SBP ≥150 mmHg or DBP ≥90 mmHg), a cut-off value of 30/15 mmHg fall is more appropriate to define OH, since BP fall after standing up largely depends on baseline BP (Fig. 1C).8,9

Different changes in blood pressure (BP) and heart rate (HR) during tilt-table tests (A-C) or during active standing tests (D, E). Green lines indicate HR. Upper red lines indicate systolic BP (SBP) and lower lines reflect diastolic BP (DBP). In (D) and (E), only SBP is shown. Orthostatic hypotension (OH) is defined as a sustained drop in SBP by at least 20 mmHg or in DBP by at least 10 mmHg (A, B). An absolute HR increase of less than 15 beats per minute or a ratio of HR to SBP (ΔOH/ΔSBP) less than 0.5 can differentiate neurogenic OH (A) from non-neurogenic OH (B). Delayed OH is a sustained decline in BP occurring after 3 minutes of standing. When supine SBP is greater than 150 mmHg, 30/15 criteria may be more appropriate for OH or delayed OH instead of 20/10 (C). Initial OH is a ‘transient’ drop in SBP by at least 40 mmHg or in DBP by at least 20 mmHg during active standing (D). Delayed BP recovery is a transient decline in BP by 20/10 during active standing, but BP recovers 30 seconds or longer after standing up and within 3 minutes (E).
Delayed orthostatic hypotension
When patients report symptoms consistent with orthostatic intolerance, orthostatic vital signs with 3 minutes of standing at clinics may not reveal any abnormalities. In such cases, delayed OH can be considered especially when there is latency of symptoms after standing up. Delayed OH is defined as a sustained drop in BP after standing for 3 minutes or long (Fig. 1C).3,8-10 It is considered a milder form of sympathetic adrenergic failure,8,10-12 and its progression to OH has been associated with α-synucleinopathies, diabetes mellitus, more severe abnormalities on autonomic reflex test, and higher mortality rates.13 However, the frequency, severity, and type of orthostatic intolerance do not differ significantly between classic and delayed OH.11,14
Initial orthostatic hypotension
Initial orthostatic hypotension (I-OH) is a ‘transient’ reduction of SBP by at least 40 mmHg or of DBP by at least 20 mmHg. The transient drop occurs in 15 seconds of standing and BP recovers in 30 seconds (Fig. 1D). It occurs more frequently in adolescents and young adults.8,15 Patients may experience transient symptoms such as lightheadedness, nausea, and blurred vision right after standing up or when making a few steps after standing.16 A mismatch between cardiac output and peripheral vascular resistance is the most likely mechanism.8 I-OH is usually a benign phenomenon. Its symptoms can disappear quickly by sitting or squatting. The usual blood pressure cuff on the arm or the wrist may not detect it. An active standing test with a continuous beat-to-beat BP measurement is required. Also, an active standing test has high specificity but low sensitivity for I-OH.
Delayed blood pressure recovery
Delayed BP recovery is characterized by an immediate and transient drop in BP when standing, followed by a gradual recovery. The recovery in SBP to <20 mmHg from the supine baseline occurs 30 seconds or longer after standing and within 3 minutes (Fig. 1E).3,10 In a large cohort study, patients on anti-hypertensive medications or with diabetic autonomic neuropathy often present transient postural symptoms of delayed BP recovery. It is associated with an increased risk of unexplained falls and fractures.17-20
Clinicians should be aware that classic OH, delayed OH, and delayed BP recovery are not mutually exclusive conditions.21 Various factors such as hydration, food-intake, ambient temperature, medications, and time spent in sitting or flat positions before standing up can affect postural hemodynamic changes.
CLINICAL PRESENTATIONS
Symptoms of orthostatic hypotension
OH can be either symptomatic or asymptomatic. Its symptoms result from hypoperfusion to the brain and other organs above the heart. Common symptoms include lightheadedness, dizziness, and falls with or without loss of consciousness.1,2,6-10 Visual symptoms can occur due to impaired perfusion to the eyes and/or the occipital lobes. Auditory symptoms such as hearing changes and tinnitus may occur because the cochleae are end-artery organs vulnerable to ischemia. Patients might report vague symptoms of fatigue, cognitive slowing, and generalized weakness that improve upon lying down.9,10,22 Pain due to ischemia in the trapezius and the adjacent muscles can occur over the suboccipital and paracervical areas making its distribution look like a coat-hanger (coat-hanger pain).9,10,23,24 Moreover, a study has also found that a 30.0% proportion of patients with OH report shortness of breath (platypnea). Its main mechanism is a ventilation-perfusion mismatch with hypoperfusion of the lung apices.9,25 In general, the most important factor that can determine if certain symptoms are due to OH is that they occur in upright positions, and improve quickly after sitting or lying down.
Blood pressure threshold for symptoms
In a study with Parkinson’s disease (PD) with OH, OH symptoms were associated with standing mean arterial pressure less than 75 mmHg, regardless of degrees of BP drop.26 The authors suggested pharmacological intervention for OH should be initiated for patients with standing mean arterial pressure below 75 mmHg. In studies with OH from various etiologies, there were no hemodynamic parameters that could predict symptoms consistently suggesting a weak correlation between symptoms and hemodynamic measurements.27,28
CLINICAL APPROACH
In-clinic monitoring
At clinics, BP and HR are measured after at least 3-5 minutes of resting in a supine position, ideally followed by measurements at 1 and 3 minutes of standing.2,6 When this method is not feasible at a clinic, criteria of 15/7 mmHg instead of 20/10 mmHg can be used for sit-to-stand measurements to diagnose OH with acceptable sensitivity and specificity.29 However, it’s unclear whether this criterion can be applied to patients with hypertension in a seated position.
At-home monitoring
Studies suggest that home BP monitoring by patients is a highly feasible and efficient screening method for OH and that it is comparable to measurements supervised by healthcare providers.30,31 To minimize the effects of drugs on BP, early morning before taking any medications is an ideal time. Patients can measure BP/HR after lying down and resting. After that, they can obtain standing BP/HR after 3 minutes of standing.9 To detect OH, an orthostatic BP/HR log for 2 weeks is encouraged. Patients may need to measure BP when they experience symptoms (if feasible) or when they change medications for OH.
Neurogenic vs. non-neurogenic orthostatic hypotension
Non-neurological diseases such as acute anemia, volume depletion, adrenal insufficiency, heart failure, thyroid disorders, end-stage renal disease with hemodialysis, medications, and severe gastrointestinal diseases can cause OH. It is crucial to differentiate neurogenic OH (N-OH) from non-neurogenic OH to find its cause and manage related symptoms. HR changes may be helpful for the differentiation. Absent or minimal responses of HR to OH usually indicate N-OH. An absolute increase of HR by less than 15 beats per minute implies a higher probability of N-OH. A relative HR change to SBP drop (ΔHR/ΔSBP ratio) less than 0.5 has very high sensitivity and specificity for diagnosing N-OH, especially in autonomic neurodegenerative diseases (Fig. 1A, B).32,33 However, HR can be affected by cardiac medications, pacemakers, cardiac ablation, and aging. Autonomic neuropathies sparing cardiac sympathetic innervation may show robust HR response to OH. The presence of N-OH does not exclude non-neurogenic components of OH. Combined non-neurogenic components, particularly medications, can aggravate OH in patients with N-OH.
Etiologies of neurogenic orthostatic hypotension
Causes of N-OH can be divided into central and peripheral diseases. Among peripheral causes, diabetic mellitus is the most common condition. OH can be easily observed in patients with diabetes.34 N-OH can be seen in acute immune-mediated diseases such as autoimmune autonomic ganglionopathy, seronegative autoimmune autonomic neuropathy, and Guillain-Barré syndrome.35 Hereditary neuropathies such as familial amyloidosis polyneuropathy36 as well as acquired neuropathies due to B12 deficiency, Sjogren disease, systemic lupus erythematosus, thyroid disease, and human immunodeficiency virus may result in N-OH. As central causes, there are spinal cord diseases (e.g., spinal cord injury, syringomyelia, transverse myelitis)6,37 and neurodegenerative diseases. Autonomic neurodegenerative diseases, collectively called α-synucleinopathies, include PD, multiple system atrophy (MSA), dementia with Lewy bodies, and pure autonomic failure (PAF) sharing pathological findings of abnormal α-synuclein accumulation.6,10,38
PATHOPHYSIOLOGICAL CONCEPTS
Arterial baroreflex and inadequate release in norepinephrine
The major pathophysiological mechanism of N-OH is an inadequate release of norepinephrine after standing up. Fluid shift and blood pooling in the dependent areas after standing up produce a cascade of hemodynamic changes, including decreased venous return, a reduction in stroke volume by up to 40.0%,39 an overall decline in cardiac output, and eventually lower BP.4,6,10,40 The mechanoreceptors of the arterial baroreflex in the carotid sinus and the aortic arch sense changes in tension of the arterial walls secondary to changes in BP. They send afferent signals to the brainstem via the vagus and glossopharyngeal nerves.4,6,40,41 The signals are processed in the brainstem in coordination with the higher-order central autonomic network.40,42 The main effector organs of the baroreflex are the blood vessels and the heart. The rostral ventrolateral medulla neurons send efferent signals to the intermediolateral cell column in the spinal cord (T1-L2) where sympathetic preganglionic nerves exit through the ventral root and form synapses with postganglionic sympathetic adrenergic fibers.10 There is also a separate efferent pathway adjusting cardiovagal tone to the heart. The activated adrenergic fibers increase release of norepinephrine (NE), a key neurotransmitter for sympathetic vasoconstriction, at the neurovascular junction. In healthy people, plasma NE levels are doubled after standing from supine levels. However, NE increase is blunted in patients with N-OH (Fig. 2).43
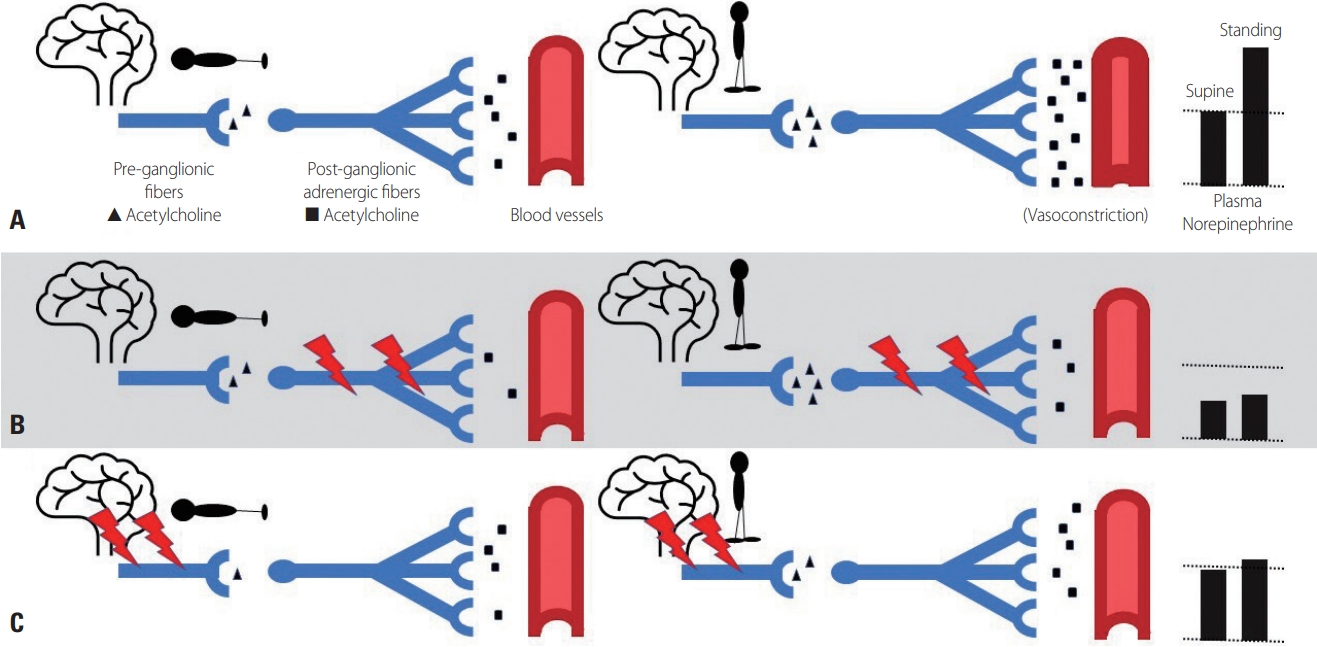
Norepinephrine (NE) changes after standing up. In a supine/resting state, there is a baseline release of NE from the post-ganglionic adrenergic fibers. After standing up, signal traffic in the autonomic ganglia increases due to the enhanced release of acetylcholine from the pre-ganglionic sympathetic fibers. It activates the post-ganglionic fibers resulting in a surge in plasma NE level and vasoconstriction (A). In post-ganglionic degeneration in pure autonomic failure, the baseline NE level is decreased. Standing up does not bring up the NE level significantly (B). In pre-ganglionic degeneration in multiple system atrophy, the baseline level is normal or slightly lower than normal. But standing up does not increase the level like normal subjects (C).
Supine hypertension
Approximately half of the patients with N-OH experience supine hypertension.10,44 Also, 30.0% of patients with PD and MSA have mild to severe supine hypertension, regardless of their age, disease duration, or stage.45 Supine hypertension is largely asymptomatic especially during daytime. But it can lead to pressure-induced natriuresis at night with subsequent nocturia and poor sleep quality. Due to the forced natriuresis, OH can get worse the following morning with more severe symptoms.44,46 Supine hypertension increases the risks of cardiovascular diseases and renal failure as longterm complications. Therefore, patients with N-OH should be regularly screened for supine hypertension, especially when starting a long-acting medication for N-OH or when reporting possibly related symptoms such as frequent awakening and nocturia.44,47 The long-term benefits of the management of supine hypertension are still under a matter of investigation. Its managements are discussed in detail on the scientific statement of the professional societies.48
Denervation hypersensitivity
Chronic reduction of NE release may cause denervation hypersensitivity in the peripheral α1-adrenergic receptors. Denervation hypersensitivity exaggerates responses of the receptors to its agonists.7,49 Denervation hypersensitivity is believed to contribute to supine hypertension and unpredictable drug responses in patients with N-OH.
MANAGEMENTS
The consensus statements for N-OH management recommend a stepwise and individualized approach. Experts suggest a 4-step treatment algorithm: a review of the patient’s medications, non-pharmacologic measures, initiation of a single pharmacologic agent, and combination pharmacologic agents.9 When feasible, factors that can aggravate OH should be corrected. Medications such as diuretics, tricyclics, peripheral α1-blockers, beta-blockers, or calcium channel blockers with vasodilating effects, and nitrates should be stopped or minimized.6,8-10 Anemia may worsen OH as well.9,50 Anemia reduces systemic vascular resistance through the induction of nitric oxide synthesis and low blood viscosity.51 If OH symptoms persist and affect daily activities after correction of the precipitating factors, non-pharmacological measures need to be considered. In milder cases, pharmacological agents can be used when non-pharmacological treatments are insufficient. But, in severe cases, both non-pharmacological and pharmacological measures may need to be started spontaneously.
Non-pharmacological managements
1. Physical counter-maneuver
Physical counter-maneuvers such as squatting, leg-crossing, thigh contraction, and bending at the waist can reduce venous pooling and increase total peripheral resistance quickly through mechanical compression of the vein.3,4,52-54 Cardiac output and BP typically increase within 3-5 seconds, ultimately maintaining cerebral perfusion pressure and extending the duration of the upright position.55
2. Salt supplementation
An increase in water and salt intake may expand the intravascular volume and reduce the severity of OH. With systemic autonomic failure, there is continued urinary sodium loss even with a low-salt diet56 indicating impaired renal control of sodium balance. Excessive urinary sodium loss can result in urinary loss of water (natriuresis) contributing to a substantial decrease in intravascular volume in patients with autonomic failure.57,58 To increase fluid volume and improve orthostatic tolerance, it is recommended to have a high salt intake (6-10 grams per day) along with increased water consumption.9,49,59 However, the recommended amount should be individualized particularly in patients with combined heart failure, liver disease, severe peripheral edema, or chronic kidney disease. Long-term effects of a high-salt diet in patients with autonomic failure are not well-studied yet.58
3. Water
To expand circulating fluid, water intake needs to increase up to 2-2.5 L of water per day.2,12,60 Gulping a big glass of water (about 500 mL) over less than 10 minutes raises plasma NE level and increases BP quickly with peak effects in 20-30 minutes.61 Patients may use this method as a rescue when they experience worsening symptoms or in the morning before their morning OH medications take effects. This pressor response lasts for 1 to 1.5 hours. It is triggered by low osmolality of water. The transient receptor potential vanilloid 4 channel receptors in the portal system sense low osmolality and trigger the osmo-pressor response. Therefore, the temperature of water seems irrelevant,62,63 although some patients occasionally report better responses to cold water. Young healthy people do not have such responses. Older people without autonomic failure may have similar responses to a lesser degree. Only patients with autonomic failure have a pronounced BP response to rapid water drinking.61,64
4. Compressive garments
Compressive garments for OH include custom-fitted elastic stockings (e.g., waist-high, thigh-high) and abdominal binder.9,10,65 Considering significant blood pooling in the splanchnic and muscular circulation, waist-high compression garments are the most effective, followed by thigh-high compression stockings.66 However, their compliance is poor, and they can be cumbersome to put on, for individuals with impaired motor coordination.9 In such cases, an abdominal binder is a good alternative.67 Patients should be instructed to apply or readjust the abdominal binder in a supine position.
5. Lifestyle modification
Activities such as prolonged standing, alcohol consumption, hot showers, and saunas should be avoided as they augment vasodilation and venous pooling.9,10,68 Additionally, high-carbohydrate foods can lead to postprandial hypotension due to shift of blood into the splanchnic circulation.69 Therefore, patients who experience worsening OH symptoms after a meal should try frequent and smaller meals. In severe cases, medications such as caffeine, acarbose, and octreotide can be considered. For example, acarbose, an anti-diabetic drug, inhibits α-glucosidase and delays the release of glucose from larger carbohydrates.49
Pharmacological managements
If non-pharmacological treatments are not sufficient, pharmacological treatments can be used in combination.12 There are two categories of medications: NE enhancers and replacers. In theory, the selection of OH drugs depends on the phenotypes of N-OH: preganglionic and postganglionic types. In post-ganglionic degeneration with limited sympathetic reserve as seen in PAF and PD (Fig. 2B), NE replacers such as midodrine and droxidopa are preferred. In pre-ganglionic degeneration with relatively intact peripheral sympathetic reserve as seen in MSA (Fig. 2C), NE enhancers such as pyridostigmine or NE reuptake inhibitors to potentiate endogenous NE release may be a better choice.49 Residual sympathetic reserve can be assessed based on resting serum NE levels, degrees of HR increase to OH (cardiac sympathetic reserve), and sympathetic adrenergic response during autonomic function tests.6,49,70 Other factors including comorbidities, degrees of supine hypertension, drug side effects, and drug prices may affect choices of drugs.
1. Midodrine
Midodrine is a short-acting α1 adrenoreceptor agonist. Desglymidodrine, its active metabolite, constricts arterial and venous vessels, thus increasing BP in a dose-dependent pattern. The half-life of desglymidodrine is approximately 4 hours, and the peak plasma level is reached in 1 hour after oral administration.71 In a randomized controlled trial of 97 patients with OH, 10 mg of midodrine increased standing SBP by 22 mmHg compared with placebo.72 Other randomized trial (10 mg dose three times per day)73 and double-blind, dose-response trial (2.5, 10, and 20 mg)71 showed benefits of midodrine in OH symptoms. The U.S. Food and Drug Administration granted accelerated approval in 1996 but requested a post-market trial.10 In a subsequent phase 4 cross-over randomization study with patients who had been on stable doses of midodrine for at least 3 months, standing time on a tilt-table test was significantly shorter with placebo, which demonstrated sustained effects of midodrine.74 Due to concern about iatrogenic supine hypertension, its last dose of the day should be away from bedtime by 4 hours.53 Its common side effects include scalp itching and piloerection.49 Other potential side effects include hypertension, tingling sensation, and urinary retention. However, midodrine has minimal central side effects because it does not cross the blood-brain barrier. Midodrine is the most used medication for OH due to its availability, minimal drug interaction, and quick onset with predictable effects. It can be used as needed for planned activities requiring prolonged standing.
2. Fludrocortisone
Fludrocortisone, which is a synthetic mineralocorticoid analog, potentiates α1 receptor activity and expands plasma volume through the urinary reabsorption of water and salt.10 The effects of volume expansion last for only a few weeks.3,9,10 It is believed that fludrocortisone makes α1 adrenergic receptors more sensitive to endogenous NE through changes in DNA transcription and protein translation. Its side effects include hypokalemia, edema, heart failure, supine hypertension, and headaches.2,75 In a cohort study of 129 patients with MSA, increased mortality was observed in those treated with midodrine, fludrocortisone, or a combination of both, regardless of disease severity.76 Additionally, fludrocortisone users had higher rates of all-cause hospitalizations, particularly among congestive heart failure patients, compared to midodrine users.77
3. Droxidopa
Droxidopa is an oral synthetic amino acid that provides exogenous NE, as it is converted to NE through decarboxylation. Since droxidopa is a NE replacer, it is more effective in patients with reduced peripheral sympathetic reserve. Low resting plasma NE levels (<219.5 pg/mL) reflecting post-ganglionic degeneration were associated with a greater pressor effect of droxidopa.78 Its phase 3 studies showed significant improvement in OH-related symptoms in the first week. However, the positive effects did not last longer.79-81 Droxidopa was granted accelerated approval in 2014, which requires post-market clinical trials to demonstrate its sustainability. Therefore, the rationale and design of the neurogenic orthostatic hypotension to assess sustained effects of droxidopa therapy study have been completed recently. Its results from the first phase of an open-label trial for 12 weeks have been published.82 Results of the double-blinded trial will follow.83 The most common adverse effects of droxidopa were headache and dizziness.81
4. NE reuptake inhibitors: atomoxetine and ampreloxetine
When norepinephrine is released, approximately 90.0% of NE is taken back to the presynaptic nerves by NE reuptake transporters (NET) quickly. Atomoxetine and ampreloxetine inhibit the NET and enhance the actions of NE on the peripheral adrenergic receptors (NE enhancer). Thus, NET inhibitors are more effective in pre-ganglionic degeneration where peripheral sympathetic reserve and baseline release of NE are still relatively intact.10 Higher resting plasma NE levels are moderately correlated with the pressor response to atomoxetine.84 A randomized controlled trial suggests that atomoxetine may serve as an alternative therapeutic option to midodrine in the treatment of orthostatic hypotension in autonomic failure.85 Although ampreloxetine showed promising results in a phase 2 study,86 a phase 3 trial in patients with α-synucleinopathies was terminated prematurely due to the lack of positive outcomes. A different phase 3 study of ampreloxetine only with MSA (NCT03750552) has been recently launched.
5. Pyridostigmine
Pyridostigmine is a reversible cholinesterase inhibitor that potentiates cholinergic neurotransmission in the autonomic ganglia, both sympathetic and parasympathetic.87,88 In a double-blind, randomized study of 58 patients with neurogenic OH, pyridostigmine alone or in combination with midodrine (5 mg) improved standing BP, particularly by increasing diastolic BP, without exacerbating supine hypertension.89 However, another double-blind crossover study found that pyridostigmine was less effective than fludrocortisone in treating OH in Parkinson’s disease.90 For individuals with severe autonomic failure, although neither pyridostigmine nor atomoxetine improved BP compared with placebo, the combination of pyridostigmine (60 mg) and atomoxetine (18 mg) increased seated BP in a synergistic manner.91 Because of its modest efficacy and small clinical sample size, pyridostigmine is currently used as a second-line or adjunctive treatment option. Additionally, side effects such as nausea, diarrhea, diaphoresis, and hypersalivation can occur because of its nonselective effects on both nicotinic and muscarinic acetylcholine receptors.10,88 Further more comprehensive research involving older individuals is required to confirm its effectiveness and tolerability.
CONCLUSION
Patients with autonomic disorders may have N-OH. To find its specific etiology and determine appropriate managements, it is important to identify symptoms of OH and differentiate N-OH from non-neurogenic OH. Symptoms of N-OH result from insufficient blood supply to organs, mainly the brain, secondary to low-standing BP. There should be relief after sitting or lying down. Heart rate changes during OH may help distinguish between N-OH and non-neurogenic OH. The main pathophysiological mechanism of N-OH is insufficient increase in NE release from the sympathetic adrenergic system to the blood vessels. The patterns of plasma NE levels differ depending on phenotypes of N-OH (pre-ganglionic vs. post-ganglionic degeneration). A stepwise approach is recommended to manage N-OH symptoms. It is important to consider combined medical conditions and correct potential contributors to OH. Before initiating pharmacological treatment, non-pharmacological approaches should be explored and tailored to the patient’s situation.
Notes
Conflicts of Interest
None.