A case of X-linked Charcot-Marie-tooth disease type 1 manifesting as recurrent alternating hemiplegia with transient cerebral white matter lesions
Article information

Abstract
X-linked Charcot Marie Tooth disease type 1 (CMTX1) is a clinically heterogenous X-linked hereditary neuropathy caused by mutation of the gene encoding gap junction beta 1 protein (GJB1). Typical clinical manifestations of CMTX1 are progressive weakness or sensory disturbance due to peripheral neuropathy. However, there have been some CMTX1 cases with accompanying central nervous system (CNS) manifestations. We report the case of a genetically confirmed CMTX1 patient who presented recurrent transient CNS symptoms without any symptom or sign of peripheral nervous system involvement.
X-linked Charcot-Marie-Tooth disease type 1 (CMTX1) is a subtype of X-linked Charcot-Marie-Tooth disease (CMTX) that is caused by the mutation at Xq13.1 of the gap junction beta 1 protein (GJB1) gene encoding connexin 32 (also known as gap junction beta 1 protein).1 Like other types of CMTX, CMTX1 manifests as symptoms of peripheral neuropathy characterized by progressive distal muscle weakness, sensory disturbance, and foot deformity. However, there have been several reports of central nervous system (CNS) involvement in CMTX1.1 We report a genetically confirmed patient with CMTX1 who presented with recurrent transient CNS symptoms without any clinical symptom or sign of peripheral nervous system involvement.
CASE
A 17-year-old male was referred to us for evaluation of recurrent alternating hemiparesis that had fully recovered within a few hours. He had initially been managed under a diagnosis of multiple sclerosis for 7 years at another hospital.
His first symptom developed at 10 years of age with sudden-onset left hemiparesis that lasted for only a few hours and was not accompanied by seizure or cognitive impairment. The second attack developed at 11 years of age with sudden-onset right hemiparesis, from which he fully recovered within a few hours. At ages of 12 and 13 years he experienced the third and fourth attacks of transient right hemiplegia and dysarthria. After each event he was serially evaluated with brain magnetic resonance imaging (MRI), which revealed recurrent nonenhanced subcortical high signal intensities in T2-weighted images that disappeared within a few days (Fig. 1).
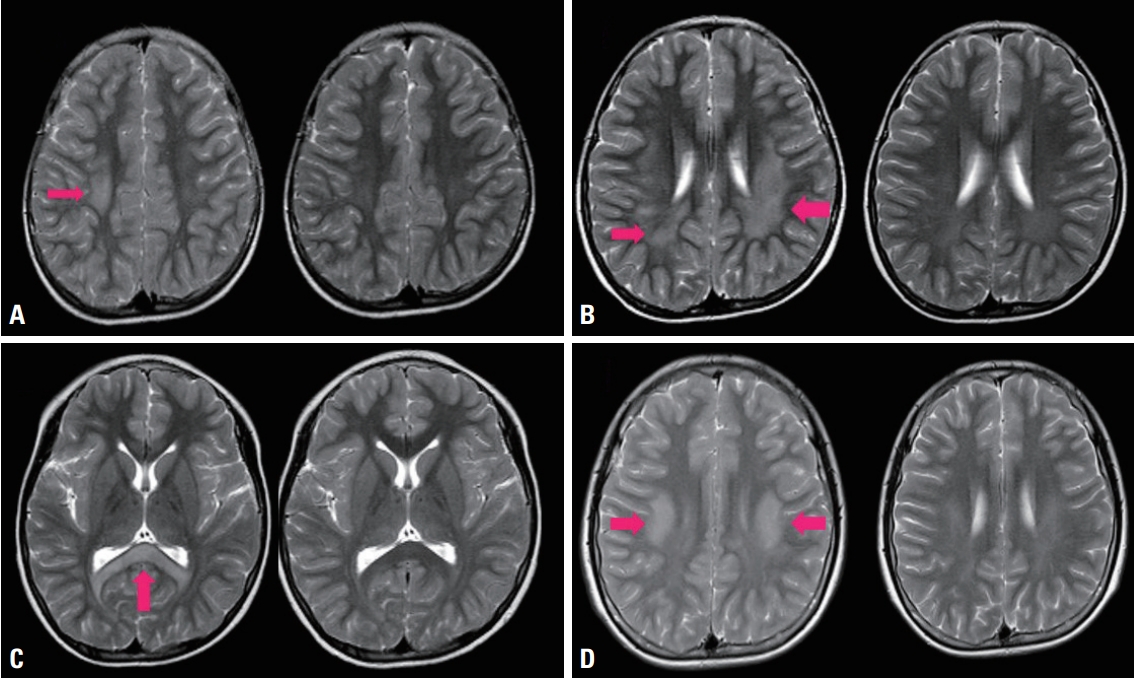
Serial brain magnetic resonance imaging findings. (A) Subcortical high-signal lesion on T2-weighted images (first attack in 2004; left image, red arrow). The signal disappeared after a few days (right image). (B-D) Similar reversible lesions (red arrows) were evident in T2-weighted images in (B) 2005, (C) 2006, and (D) 2011.
A neurological examination revealed no remarkable abnormalities, including in cognitive function, cranial nerves, motor power, muscle tone, sensory function, or deep tendon reflexes. Pathological reflexes and musculoskeletal deformities were not observed. Because somatosensory evoked potential investigations strongly suggested the presence of peripheral neuropathy (Fig. 2), nerve conduction studies (NCSs) were performed, which revealed mixed sensorimotor polyneuropathy of the upper and lower extremities (Table 1).
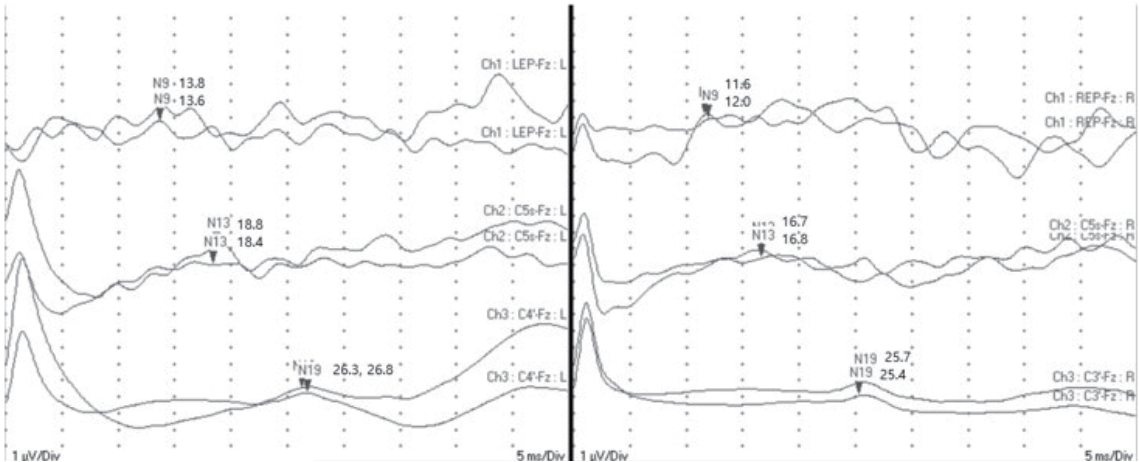
Somatosensory evoked potentials of the median nerve showed prolonged absolute latencies to N9, N13, and N19 with normal interpeak latencies, suggesting a peripheral conduction defect.

Findings of NCSs of the patient
Under a clinical impression of CMTX, which was supported by recurrent white-matter lesions on MRI and evidence of peripheral polyneuropathy on NCSs, we decided to test for mutation in GJB1, which is the most common causative gene of CMTX. Sequence analysis of GJB1 revealed a duplication of nine bases (c.572_580dup, [p.Thr191_Phe193dup]), which had previously been reported as a pathogenic mutation.2 We also examined his mother, and noted that she had pes cavus deformity and depressed deep tendon reflexes. Her NCS results also indicated sensorimotor polyneuropathy, and a DNA study showed that she carried the same mutation.
DISCUSSION
CMTX1 is associated with mutation in GJB1, which encodes connexin 32 (Cx32). Cx32 forms gap junctions in noncompact myelin produced by myelinating Schwann cells. Cx32 is also expressed in oligodendrocytes, explaining the potential of CNS involvement.3 CNS symptoms can manifest as transient limb weakness or dysarthria, and they are associated with nonenhanced deep white-matter lesions on MRI, which improve in follow-up MRI.4 In the present case, recurrent transient CNS symptoms such as hemiplegia and dysarthria were temporarily coincident with the MRI findings. A previously reported case of the same mutation (p.Thr191_ Phe193dup) presented with peripheral neuropathy but without central manifestiation.2 There has also been a report of the mutation at a similar location resulting in CNS manifestaions.5 However, it is unclear why CNS symptoms manifest in only a few patients, and so further studies are required to clarify the genotype–phenotype relationship in CMTX1.
It is notable that the present case did not show any clinical symptom or sign of peripheral neuropathy, which made a correct diagnosis more difficult over a long time period. In addition, physical signs of peripheral neuropathy manifested in the patient’s mother who carried the same mutation, while they did not in her son. This is also considered quite unusual because males usually present with more-severe symptoms and signs of peripheral neuropathy than do females in CMTX1, which is an X-linked dominant disease.
As observed in the present case, CMTX1 can be easily misdiagnosed as CNS demyelinating disorders, because a few patients may present with recurrent hemiplegia and dysarthria with supporting evidence on brain MRI.6 It is therefore advisable to include CMTX1 in the differential diagnosis of CNS demyelinating disease, especially when a patient presents with recurrent CNS symptoms associated with nonenhanced white-matter lesions on brain MRI, even in the absence of clinical evidence of peripheral neuropathy.
Notes
Conflicts of Interest
The authors have no conflicts to disclose.
Acknowledgements
This study was supported by a 2020 research grant from Pusan National University Yangsan Hospital.