Likely pathogenic FIG4 related amyotrophic lateral sclerosis patient who correlated with clinical, imaging and neuropsychological studies
Article information

Abstract
Amyotrophic lateral sclerosis (ALS) is a progressive neurodegenerative disorder with numerous causes that include genetic factors. Efforts to reveal the genetics of ALS have identified several candidate genes that are associated with familial and sporadic ALS. Here we report a Korean ALS patient who showed prominent upper motor-neuron-related symptoms with marked brain atrophy and neuropsychological deficits. The findings were highly suggestive of ALS in a patient with a likely pathogenic FIG4 variant.
Amyotrophic lateral sclerosis (ALS) covers a group of neurodegenerative diseases. About 5-10% of ALS cases are familial, and 19 genes are known to be causative factors. The most-attributable gene in Caucasian populations is the C9orf72 gene, which accounts for about 40% of familial ALS cases and 7% of sporadic ALS cases.1 SOD1 genes account for a further 15-20%, followed by FUS, OPTN, VCP, and UBQLN2 genes. In contrast, among Asian populations (including Korean) C9orf72 is known to be a very rare cause, suggesting the presence of racial differences.2 We report a patient with ALS associated with a likely pathogenic FIG4 variant who presented with clinical symptoms and imaging findings that are in accordance with recent literature on FIG4-related ALS.
CASE
A 69-year-old woman complained of stiffness and weakness in her left limb that had first appeared 6 months previously. At the initial visit she complained of right hand weakness, difficulty using chopsticks, and recent memory impairment. The patient had no family history of significant neurological disease. A neurological examination showed mild dysarthria as well as tongue atrophy and fasciculation. A motor examination revealed weakness in her left and right arms, with Medical Research Council scores of 3 and 4, respectively. Her tendon reflexes were abnormally hyperactive, with positive pathological reflexes. A nerve conduction study showed decreased compound action potentials in the motor nerves, and needle electromyography showed active denervation potentials in both cervical and lumbar spine-related muscles. Laboratory tests including autoimmune, very-long-chain fatty acid, and other cerebrospinal fluid tests produced no specific findings. She was finally diagnosed as clinically probable ALS according to the revised El Escorial criteria.
Her score on the revised Amyotrophic Lateral Sclerosis Functional Rating Scale (ALSFRS-R) at the time of diagnosis was 21 points, but this deteriorated rapidly with a monthly decrease of more than 1 ALSFRS-R point. The patient was aware of the memory decline from the time of the weakness, and her husband reported that her personality had changed significantly. The Korean version of the Mini Mental State Examination showed a marked decline in cognitive function, with a score of 19/30. Her Clinical Dementia Rating was 0.5/4 for the sum of boxes. The second edition of the Seoul Neuropsychological Screening Battery, the digit span test, the Korean version of the Boston Naming Test, the frontal contrast test (contrasting programming & Go-No-Go test), Fistedge-palm, Stroop, and Luria loop tests were applied, which revealed impairments in attention and naming as well as the frontal lobe dysfunction. In accordance with the above neuropsychological findings, brain magnetic resonance imaging revealed significant cortical atrophy even after considering age-related changes (Fig. 1).
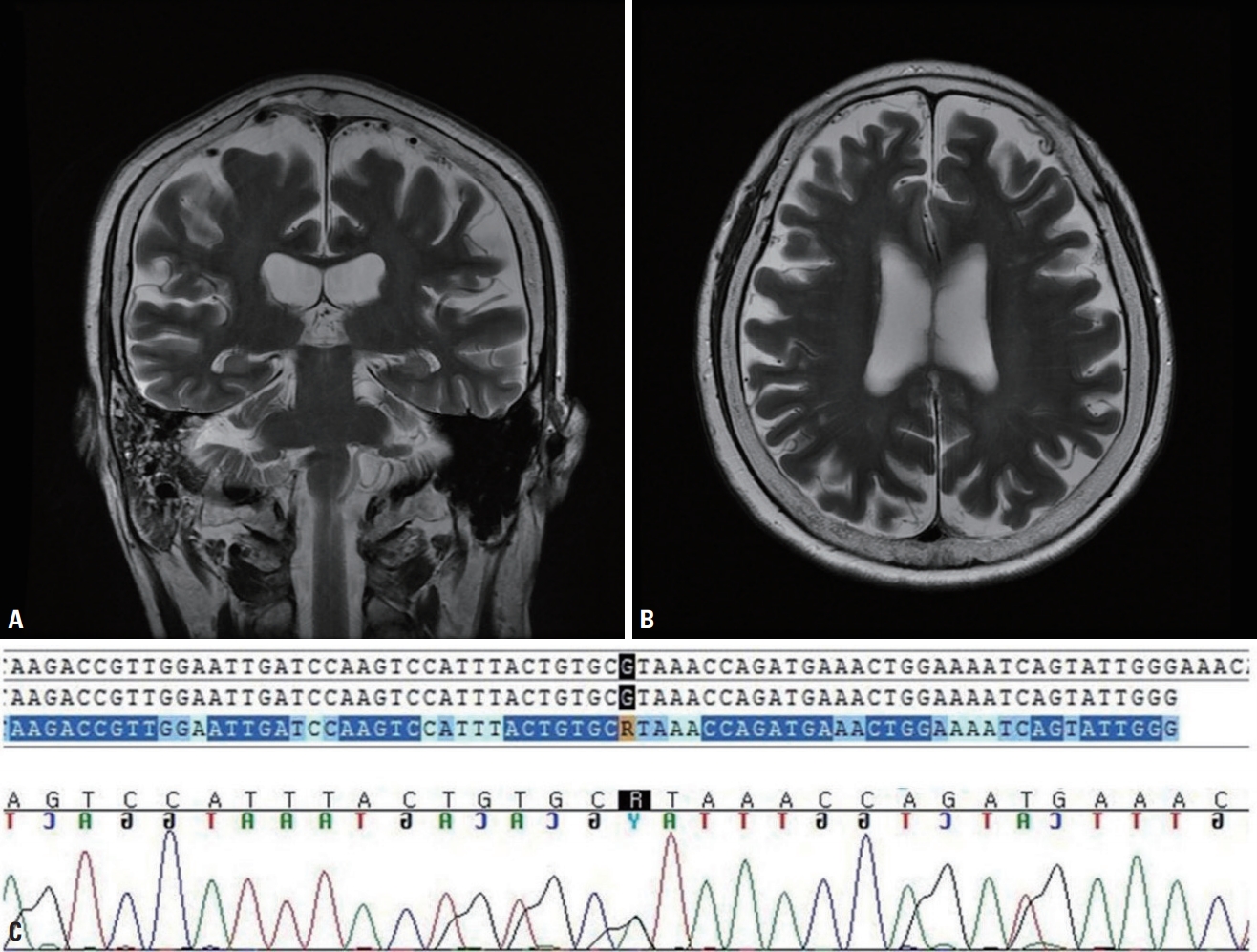
(A) Coronal and (B) axial brain magnetic resonance imaging revealed frontal dominant cortical atrophy. Sanger sequencing of the patient revealed a missense mutation in the FIG4 gene (c.2147G>A, R716H) that was predicted to be deleterious in in-silico analyses (C).
The patient was finally diagnosed clinically as ALS with acute cognitive decline, and so a targeted ALS gene panel test including C9orf72 was performed. This test identified a missense variant of c.2147G>A, R716H in the FIG4 gene. Although we were unable to perform any functional tests, this missense mutation is not reported in the 1,000 Genomes or ExAC database, and it was predicted to be deleterious causing significant protein changes in the in-silico analyses performed using SIFT and PolyPhen. Based on the American College of Medical Genetics and Genomics (ACMG) standards, we classified this variant as likely pathogenic.
DISCUSSION
Since the discovery of the first SOD1 gene mutation in 1993, 19 genes have been identified as causative genes in familial and sporadic ALS. The FIG4 gene was first known as a causative gene in type-4J Charcot-Marie-Tooth disease,3 and recent studies have shown FIG4 to be the causal gene of familial and non-familial ALS,4 Interestingly, FIG4 is also known to cause Yunis-Varon syndrome, familial epilepsy, and polymicrogyria.5 The FIG4 gene was found to account for about 3% of ALS cases (ALS11) in a recent European cohort.6
The pathomechanism of FIG4 has not yet been elucidated, but currently it is known to contribute to encoding various phosphoinositides that regulate the trafficking of vesicles to the trans-Golgi network. It is also closely involved with the degeneration of motor neurons. ALS11 is a rare autosomal dominant form of ALS associated with heterozygous mutations of FIG4, and is known to show incomplete penetration even in a single family.5 The family history was unfortunately not clear in our case, possibly due to the incomplete penetration rate of the FIG4 gene or there being a de-novo variation over multiple generations; however, experimental studies are necessary to confirm this. According to the recent literature, patients with FIG4-related ALS are known to show prominent upper motor symptoms, such as marked atrophy of the brain that typically involves the frontal and parietal lobes with enlarged ventricles [6]. These clinical and neuroimaging findings are supported by animal experiments showing extensive neuronal degeneration typically in the motor cortex rather than in the spinal motor neurons.3 In accordance with the above clinical and experimental findings, our patient also had diffuse brain atrophy that was correlated with the neuropsychological findings.
Approximately 33-51% of patients with ALS are known to show mild cognitive impairment.7,8 Our patient also showed impairment in attention and frontal/executive functions, while her memory and visuospatial functions were relatively preserved compared to the normal levels in aged subjects. These findings are consistent with the cognitive decline previous observed in FIG4-related ALS studies.5,6
To date, only 17 patients with FIG4 mutations have been reported worldwide.4,6 Among them, 76% had missense mutations and these reported mutations were distributed in various domains, without any hotspots. Our case also showed a likely pathogenic missense variant that is consistent with the previous findings. The main limitation of the present case is that we were unable to perform any experiments to prove its pathogenicity, and so relied and presumed to be a de novo mutation relying on a negative family history. However, such cases have not been reported in the Korean Reference Genome Database, and so the particular variant was classified as likely pathogenic according to the ACMG criteria. Furthermore, clinical and neuroimaging findings were consistent with the findings of previous studies of ALS related to FIG4 mutations.
In conclusion, we have report an ALS patient with a high probability of carrying a likely pathogenic FIG4 variant. More cases are warranted to understand the clinical and genetic characteristics of FIG4 in ALS patients.
Notes
The authors have no financial conflicts of interest.