Leigh syndrome (LS) is a mitochondrial disorder with a wide range of clinical manifestations including developmental delay, hypotonia, ataxia, dystonia, and ophthalmologic abnormalities such as nystagmus and optic atrophy.1 The onset generally occurs by the age of 2 years, and patients often die by the age of 3 years. LS is one of the most common mitochondrial disorders in children, and adult-onset LS is rare. Adult patients can present with typical features of LS or with more diverse symptoms.2
More than 75 disease genes have been identified for LS, indicating that it is heterogeneous. One of the pathogenic mutations is m.9176T>C, which encodes adenosine triphosphate synthase subunit 6 of complex V. The association between LS and this mutation was first described in 1997, and subsequently confirmed and explored.3-6 The present case report describes a case of adult-onset LS due to m.9176T>C mutation, with cortical involvement.
CASE
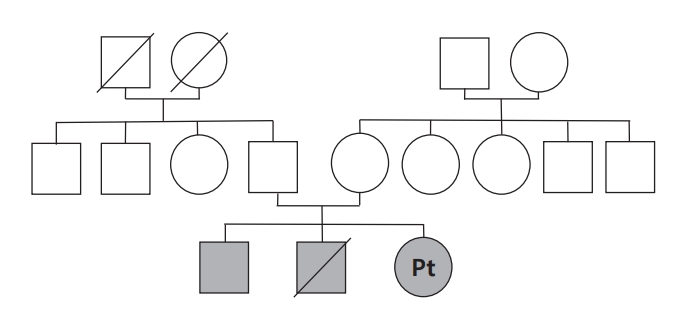
A 24-year-old female presented with diplopia and ptosis of both eyes with a 1.5-month history. She had a family history of diplopia and gait disturbance (Fig. 1). One of her two older brothers developed diplopia at 12 years old and gait disturbance during his 20s, both of which showed spontaneous remission within a few months from onset. Her other older brother died at 5 years old, a few weeks after he had developed gait disturbance. A neurologic examination of the patient indicated adduction limitation in both eyes, anisocoria with sluggish light reflex in the right eye, bilateral ptosis, and mild swaying gait without limb ataxia. Lactate and pyruvate levels were mildly elevated, at 1.7 mmol/L and 1.42 mg/dL, respectively, but other laboratory findings at admission including of a cerebrospinal fluid (CSF) analysis were unremarkable.
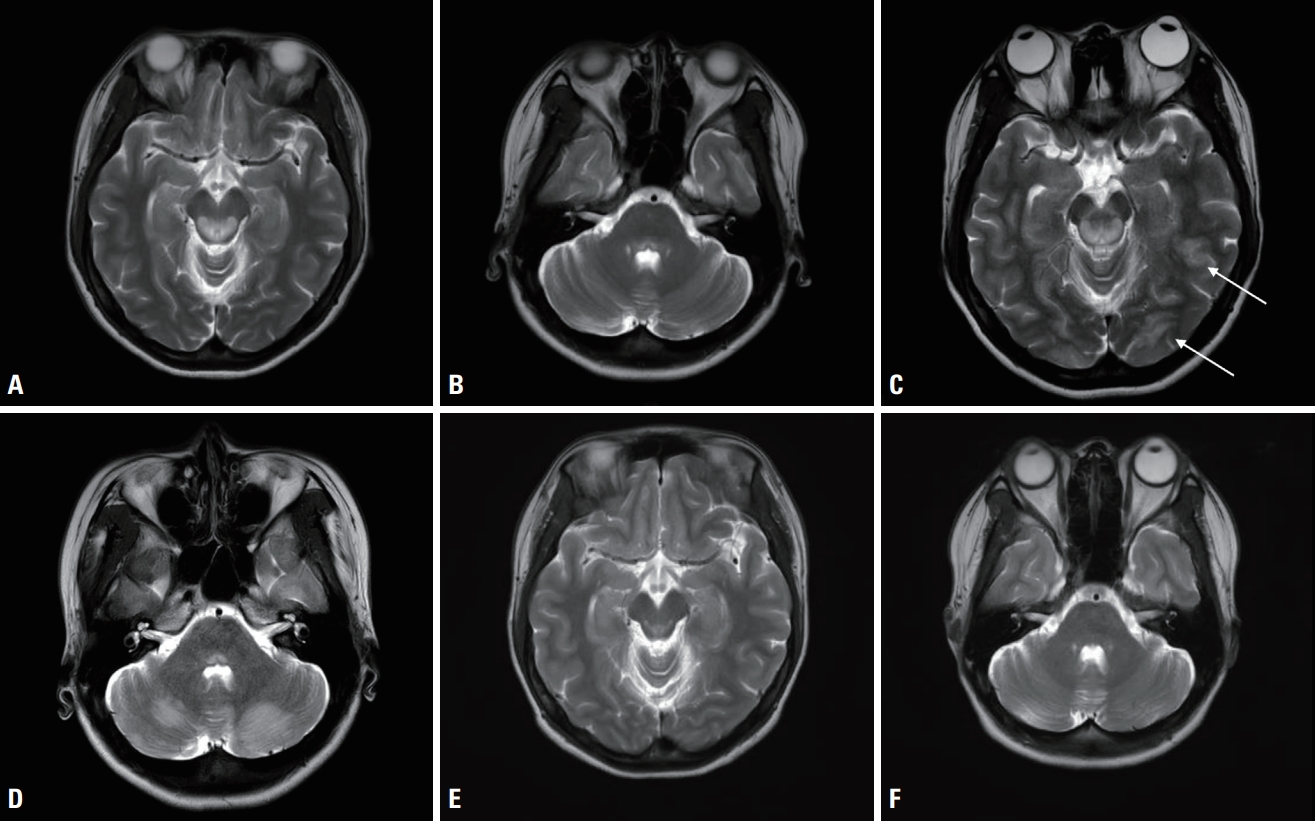
Magnetic resonance imaging (MRI) revealed a symmetric lesion in the dorsal midbrain and the pons, lateral medulla, and medial thalamus, and an asymmetric lesion in the left occipital and temporal sulci (Fig. 2A, B). High-dose thiamine was initially empirically administered to due to the possibility of Wernicke encephalopathy. On the 4th day after admission, the clinical course progressed to generalized tonic-clonic seizures, which indicated an urgent need for anticonvulsant therapy. She also complained of blindness in both eyes. After three serial seizure events, her mentality diminished to a stupor. Follow-up MRI showed new T2-weighted hyperintensities in the cerebellum and prominent signal changes in both occipital cortices (Fig. 2C, D). Electroencephalography indicated generalized continuous slow waves. Lactate peak was identified in magnetic resonance spectroscopy.
High-doses of vitamins A, B, C, and E with acetylcarnitine and coenzyme Q10 were administered. She was also treated with empirical methylprednisolone pulse therapy for 3 days, which was tapered to oral prednisolone. Her mentality became alert after being treated with this regimen for 4 days, with the treatment being continued. On day 40 after admission she was discharged with remarkable improvement of diplopia, while the vitamin cocktail therapy without steroids was maintained.
The LS diagnosis was confirmed by whole mitochondria DNA sequencing, which analyzes 372 nuclear and 37 mitochondrial DNA associated with mitochondrial components. The m.9176T>C mutation with 100% homoplasmy was found. The patient had fully recovered within a few months after discharge, and MRI conducted 16 months after discharge demonstrated almost complete regression of the previously identified lesion (Fig. 2E, F).
DISCUSSION
The criteria for a stringent LS diagnosis defined in 1996 and amended in 2015 by Rahman and colleagues requires characteristic neuropathology or neuroradiology that must be accompanied by progressive neurodegeneration with (1) clinical evidence of brainstem or basal ganglia dysfunction, (2) intellectual and motor developmental delay, and (3) abnormal energy metabolism indicated by a severe defect in oxidative phosphorylation or pyruvate dehydrogenase complex activity, a molecular diagnosis in a gene related to mitochondrial energy generation, or elevated serum or CSF lactate.7 Our patient presented ophthalmoplegia correlated with diffuse brainstem lesion and was found to have the m.9176T>C pathogenic mutation, which satisfied the criteria for an LS diagnosis.
What is remarkable in our patient was the lesion identified by MRI including the bilateral occipital cortex and the patient presenting a correlated symptom of cortical vision loss. MTND5 and MTND3 mutations underlie rare clinical presentations of LS/mitochondrial encephalopathy, lactic acidosis, and stroke-like-episode overlap syndrome.7 Patients with these mutations occasionally show cortical lesions on MRI.8 However, we are not aware of any previous report of cortical lesions and symptoms in patients with late-onset LS who carry the m.9176T>C mutation. One case report described a 26-year-old female who presented with diplopia, was complicated by generalized seizure, and was confirmed to have the same mutation as our patient.6 Although MRI of the previous report revealed an occipital subcortex lesion, the present case presented with an occipital cortex lesion and cortical vision loss as well as a generalized seizure. This implied the possibility of cortical involvement in LS by the m.9176T>C mutation as well as by the MTND mutation.
The present case is also of value due to the excellent prognosis. The patient deteriorated on the 4th day, reporting blindness and seizure, followed by stupor. After administering a vitamin cocktail, antiepileptic drug, and empirical steroid for 4 days, she became alert and recovered her vision. After being discharged, she showed further continuous improvement of ophthalmoplegia, which was her main complaint. Vitamin B is recommended for treating several mitochondrial diseases because it serves as a cofactor for many mitochondrial reactions and is a precursor of reducing agents.9 As antioxidants, vitamin C, vitamin E, and acetylcysteine are also advocated for use in some mitochondrial diseases since they may reduce oxidative stress caused by mitochondrial dysfunction and mitigate DNA, lipid, and protein damage within cells.9 The vitamin cocktail prescribed in the present case contained vitamins A, B, C, and E, and acetylcarnitine and coenzyme Q10.
As well as vitamin cocktail therapy, intravenous steroids might also work. The effect of immunomodulatory therapy in LS was suggested by a case report of a patient with LS who was responsive to intravenous immunoglobulin and plasmapheresis.10 There was another case report where the rapamycin analogue everolimus led to radiologic and clinical improvement in a patient with LS.11 Based on the study that found that 61% of acute exacerbations resulting in hospitalization of patients with LS were caused by infection, and other literature suggesting a temporal relationship between infection and the onset of mitochondrial disease symptoms, an immune response is thought to be one of the triggers for disease onset.12 Although the exact mechanism is under investigation, cell necrosis via energy deficiency in mitochondrial disorder may lead to the release of inflammatory cytokines or cellular mediators of immunity and activate immune and inflammation pathways. These pathways might be affected by steroids and are thought to be future candidates for mitochondrial disorder research. There have actually been increasing amounts of clinical and preclinical studies that found immune targeting interventions to be helpful in various mitochondrial diseases.12
We conducted thorough testing to detect mutations of the genes related to mitochondrial components, including not only the well-known mutation but also all the mitochondrial and nuclear DNA that encode the mitochondrial component. This means that other genetic factors that might affect the phenotype were excluded.
In conclusion, the present case suggests that the m.9176T>C mutation can be a cause of LS with cortical involvement and that immunomodulatory treatment and vitamin cocktail therapy are possible options for mitochondrial disorder.