AGel amyloidosis, also known as the Finnish type of hereditary gelsolin amyloidosis, gelsolin amyloidosis, and Meretoja syndrome, is an autosomal dominant inherited disease caused by gelsolin mutations that was initially reported in a Finnish family.1,2 Patients with AGel amyloidosis typically manifest as the clinical triad of lattice corneal dystrophy, cutis laxa, and progressive cranial neuropathy.1 Here we report a family who presented with progressive cranial and bulbar palsies and was diagnosed with AGel amyloidosis using whole-exome sequencing (WES).
CASE
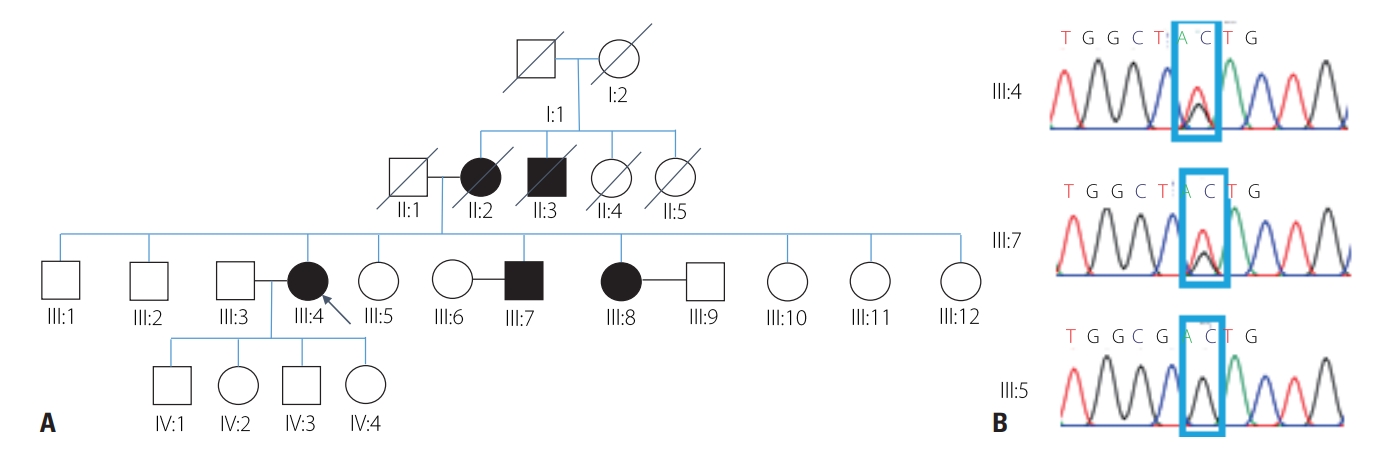
The proband was a 71-year-old female with progressive dysarthria, dysphagia, and facial palsy with a 15-year history. Facial twitching, facial weakness, and dysarthria developed at the age of 55 years, and the symptoms had slowly progressed. She denied any limb weakness, sensory change, or respiratory problem. She underwent cataract surgery at 65 years of age. Her familial history indicated that her mother, a maternal uncle, a brother, and a sister had also experienced dysarthria, bilateral ptosis, and facial palsy since their 50s (Fig. 1A). All members of her family were ethnic Koreans.
A neurologic examination revealed facial diplegia with fasciculation, bilateral ptosis without external ophthalmoplegia, dysarthria, and tongue atrophy with fibrillation. Mild neck flexion weakness was observed, but limb muscles showed normal power. Sensory and cerebellar functions were well preserved. Deep tendon reflexes were normoactive and symmetrical, and pathologic reflexes were not evoked. She had lax skin, especially on her cheek (Fig. 2).
Routine laboratory examinations including complete blood count, liver and renal functions, urinalysis, and serum creatine kinase level produced normal findings, as did a cerebrospinal fluid examination, brain magnetic resonance imaging, and routine nerve conduction studies of the extremities. Electromyography revealed abnormal spontaneous activity in the hypoglossus muscle and giant motor-unit action potentials in the hypoglossus, masseter, and orbicularis oculi muscles, but the findings in limb muscles were unremarkable. The video fluoroscopic swallowing test showed an abnormal oral phase and vallecular pooling. She also complained of eye dryness, and an ophthalmologic examination showed superficial punctate keratopathy due to contact keratitis. However, lattice corneal dystrophy was not observed despite performing repeated slit-lamp biomicroscopic examinations. Because of worsening bilateral blepharoptosis and pterygium, the frontalis sling procedure with blepharoplasty was performed at the age of 70 years. An audiologic evaluation showed normal hearing function.
WES performed to identify the genetic cause of the disease revealed a heterozygous missense mutation of GSN (NM_000177.5: c.640G>T; NP_000168.1: p.Asp214Tyr, rs121909715), which is a known pathogenic variant.3 We also performed a sequence analysis of her family members, and found the same variant in clinically affected members (Fig. 1B).
DISCUSSION
Gelsolin is a calcium-sensitive protein that binds to actin fibrils. There are two isoforms of gelsolin: 1) intracellular gelsolin is a regulator of actin filaments and 2) the secreted form functions as a scavenger of released actin. A GSN mutation leads to aberrant processing of the secreted form of gelsolin, and leads to abnormal proteolysis and the formation of amyloids in various tissues.4 This results in the development of various systemic features, including neurologic symptoms (facial palsy and ptosis, polyneuropathy, carpal tunnel syndrome, and dysarthria), ophthalmologic symptoms (corneal lattice dystrophy, impaired vision, cataract, and corneal ulcers), and dermatologic symptoms (cutis laxa and dry skin).5 The ophthalmologic manifestations are the most prevalent, followed by cutis laxa and cranial neuropathy.2 Ophthalmologic symptoms were initially the most frequent in a Finnish cohort aged in the late 30s, with facial diplegia, dysarthria, and polyneuropathy being less common and developing later.2
Two missense mutations (p.Asp214Asn and p.Asp214Tyr) are the most common and recurrent mutations of GSN, and they share similar clinical features.3 Other mutations have also been reported, including p.Gly194Arg, p.Asn211Lys, Pro459Arg, Met544Arg, Ala578Pro, and Glu580Lys.6 There have been two previous reports of Korean families with AGel amyloidosis, both of which shared the same mutation (p.Asp214Tyr) as the present case.7,8
Progressive facial and bulbar palsies were the predominant neurologic problem in the present case. Although other neurologic problems such as peripheral sensorimotor polyneuropathy, hearing disturbance, carpal tunnel syndrome, and ataxia are also frequent in AGel amyloidosis, we were not able to obtain any clinical or electrodiagnostic evidence for such problems in the present case.2 It is also noteworthy that corneal lattice dystrophy, which is the most prevalent and earliest problem in AGel amyloidosis, was not observed in the present case despite performing repeated slit-lamp examinations. This finding is unique because all members of the other previously reported Korean family with AGel amyloidosis showed corneal lattice dystrophy in slit-lamp examinations.8
The present case and many previous reports in the literature strongly suggest that AGel amyloidosis should be included in the differential diagnosis of progressive bulbar palsy from a neurologic perspective. Careful familial history-taking and systemic examinations are essential to diagnose this rare inherited disease.