Hereditary spastic paraplegia (HSP) is a clinically and genetically heterogeneous group of neurodegenerative diseases characterized by insidiously progressive weakness and spasticity of the lower extremities. The marked genetic heterogeneity of HSP is reflected by reports of many different associated gene loci, involving autosomal dominant, autosomal recessive, X-linked, and maternal inheritance. HSP with a thin corpus callosum (HSP-TCC) is the most common form of autosomal recessive HSP and is caused by mutations in SPG11/KIAA1840 gene on chromosome 15q.1 At least 127 mutations in SPG11 have been discovered and most patients exhibit slowly progressive spastic paraplegia, cognitive impairment and a thin corpus callosum.2 Some patients also show dysarthria, upper limb spasticity, distal amyotrophy, cerebellar ataxia, early-onset levodopa-responsive parkinsonism, and polyneuropathy.3 We describe the clinical and neuroradiological features of a family with HSP-TCC caused by a novel homozygous mutation in SPG11.
CASE
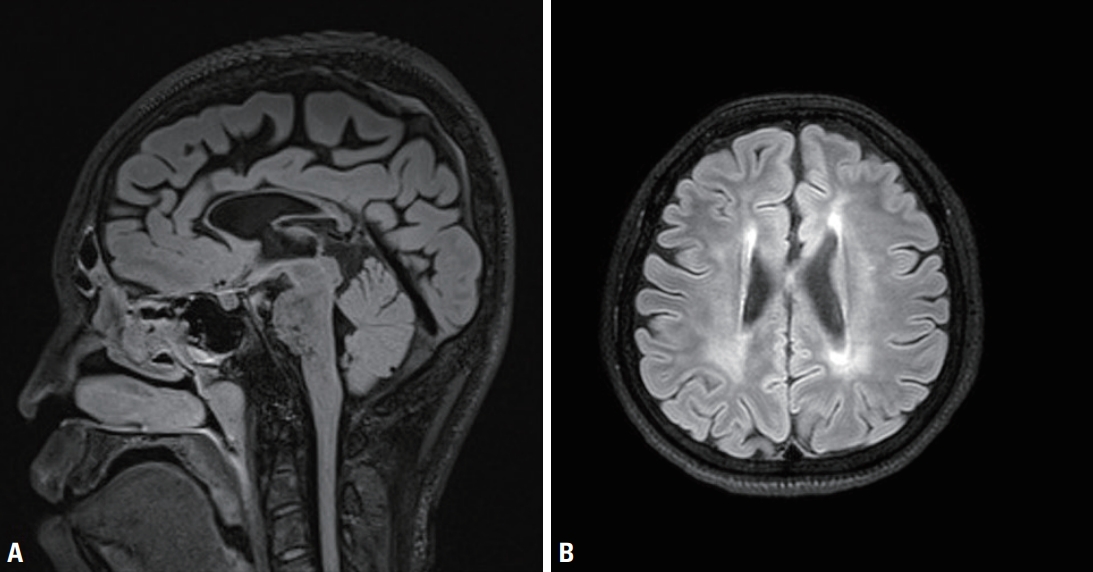
A 17-year-old male presented with gait disturbance and weakness of the lower limbs. His gait problem had first appeared when he was 12-yearold, since when it had slowly progressed. A neurological examination revealed foot drop, spasticity, hyperreflexia, and pathological reflex in both legs, but no abnormality in the upper limbs. Eye movement, cranial nerve, cerebellar. and fundoscopic examinations produced normal findings. His academic level at high school was normal, and his Korean Mini-Mental Status Examination (K-MMSE) score was 28/30. The findings of a nerve conduction study and electromyography were unremarkable. Spine magnetic resonance imaging (MRI) produced unremarkable findings, but brain MRI revealed thinning of the corpus callosum (Fig. 1). Both of his parents had normal gaits, but his 20-year-old sister had first noticed a slowly progressive gait disturbance when she was 16-year-old. She also had weakness and spasticity in the lower limbs with neurological findings that were similar to those in the proband. Also, brain MRI showed thin corpus callosum. She exhibited slow and slurred speech and mild cognitive impairment (K-MMSE score of 25).
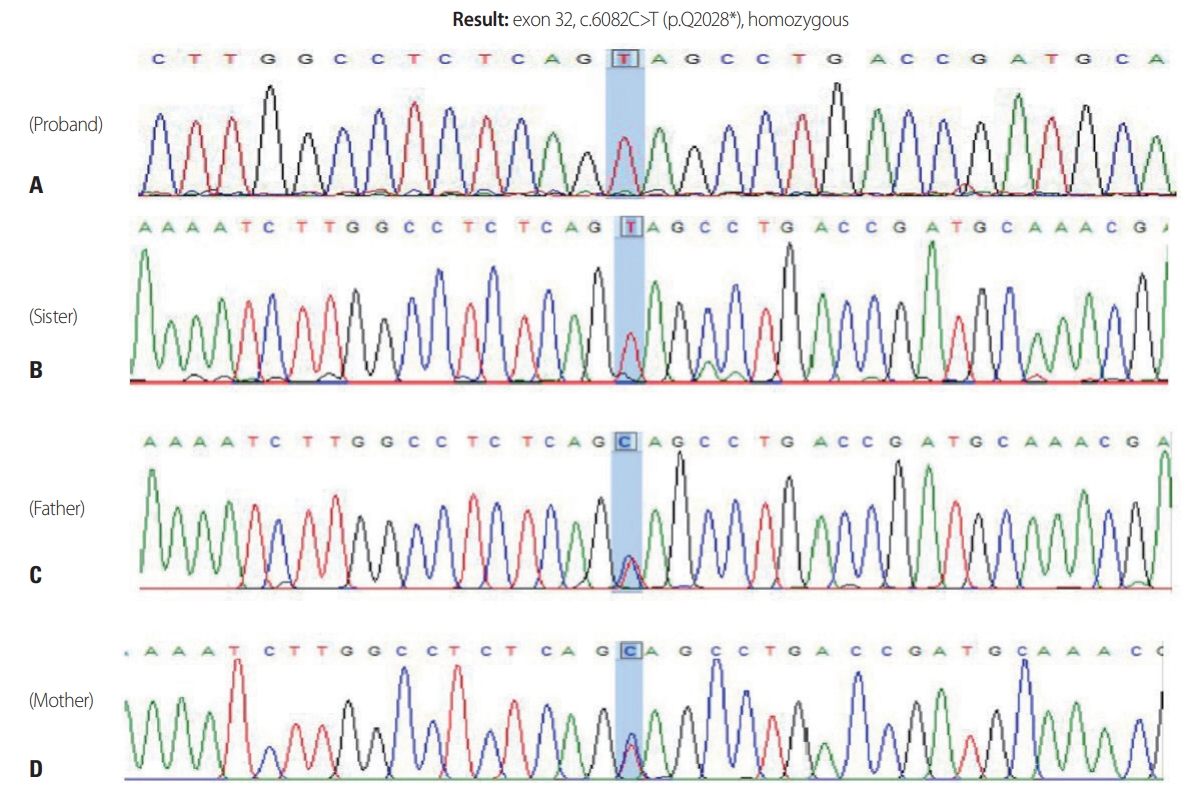
Genetic analysis by polymerase chain reaction-Sanger sequencing of all exons of SPG11 and flanking regions of each exon revealed a homozygous nonsense mutation (c.6082C>T) in exon 32. Mutation detection in SPG11 was performed in all family members. The sequencing analysis showed that both parents carry a heterozygous SPG11 c.6082C>T mutation, while the sister showed the same homozygous mutation as the proband (Fig. 2).
DISCUSSION
We have identified a novel homozygous nonsense mutation in exon 32 of SPG11 in a Korean family. Truncating mutations including nonsense and frameshift mutations are typically found in cases with HSP-TCC, as in the present case. Although the p.Q2028* mutation has not been reported previously to the best of our knowledge, it is considered pathogenic, considering that mutations that truncate the C-terminal more than this one have been reported to be pathogenic. This mutation leads to a reading frameshift and results in the premature termination of the protein, thereby truncating amino acids from the C-terminus. Our sequencing showed that both parents carried the heterozygous SPG11 c.6082C>T mutation, while the sister carried the same homozygous mutation as the proband. Both parents were clinically unaffected and our patients had inherited both of the parental disease haplotypes.
HSP is a rare neurodegenerative disorder characterized by the degeneration of corticospinal tract motor neurons and resulting in progressive weakness and spasticity of the lower extremities, which may be combined with additional neurological or non-neurological manifestations.4 HSP is classified by the phenotype, the trait of inheritance, and the mutated gene. SPG11 mutation is the most common cause of HSP-TCC and the most frequent cause of autosomal recessive HSP.1 Previous studies have found that HSP caused by SPG11 mutations is characterized by progressive spasticity with an early onset, cognitive impairment, peripheral neuropathy and a thin corpus callosum. Mild or moderate mental impairment and progressive cognitive decline are common,5 and there may also be upper limb involvement and nystagmus. A few patients additionally present with ataxia or dopa-responsive parkinsonism.6 It is particularly interesting that white matter abnormalities, which are present in more than 50% of cases, may lead to a diagnosis of leukodystrophy in patients with SPG11 mutations. Abnormal findings on electromyography and pseudobulbar dysarthria provide evidence of lower motor neuron involvement in the majority of patients.7 A subtype of SPG11 is KjellinŌĆÖs syndrome, which is characterized by spastic paraplegia, mental retardation, amyotrophy, and macular dystrophy.3
HSP-TCC is due to mutations in the KIAA1840 gene on chromosome 15q, which encodes for spatacsin. The function of spatacsin is unknown, but it is ubiquitously expressed in the nervous system, particularly in the cerebellum, cerebral cortex, and hippocampus. Spatacsin co-localizes with protein-trafficking vesicles, the mitochondrial surface, and microtubules and interacts with members of the adaptor protein complex and with spastizin.8 Frameshift, nonsense, and splice mutations in the gene for spatacsin have been identified in many families with SPG11-associated HSP-TCC, suggesting a loss of function. Spatacsin loss of function appears to produce lysosome depletion via abnormal lysosomal lipid clearance, which affects the initiation of lysosomal tubulation.9,10
We have characterized the phenotype and identified the genotype of HSP-TCC patients. Our findings suggest that novel homozygous mutations in SPG11 are associated with HSP-TCC. The association of a thin corpus callosum with cognitive impairment seems to be the best phenotypic predictor of SPG11 mutations.