INTRODUCTION
Assessments of patients with muscle weakness include a detailed neurological examination, electrophysiological studies including nerve conduction studies and needle electromyography, blood tests, and imaging studies. A muscle biopsy is needed when the results of the assessment processes indicate that the muscle weakness might be caused by a muscle disease. Muscle pathology findings are helpful for differentiating myopathies of various causes, since most types of myopathy have the common clinical manifestation of simple muscle weakness regardless of variations in their etiologies. Knowledge of the muscle pathology will also be helpful for differentiating neurogenic and myopathic muscle weakness.
Muscle pathology findings can provide a considerable amount of information about the pathological processes causing muscle weakness. The principal findings include muscle fiber atrophy and hypertrophy, necrosis and regeneration, inflammatory infiltration, and abnormal inclusions within muscle fibers. However, it should be remembered that such findings are not always specific to a particular disease, and can actually confuse the diagnosis in many cases. Genetic testing can sometimes replace a muscle biopsy, such as when the clinical features are sufficiently characteristic of a specific diagnosis, including myotonic dystrophy and facioscapulohumeral muscular dystrophy. Recent developments in next generation sequencing have led to genetic tests being directly applied to more cases of inherited myopathies, because whole exome sequencing or myopathy gene panel sequencing makes a genetic diagnosis easier by providing broad genetic data. However, clinical and pathological information is still needed to distinguish true pathogenic variations from the huge amount of genetic data. Furthermore, knowledge of the muscle pathology becomes more valuable when inflammatory myopathy is suspected. Recent improvements in the understanding of pathological features have changed the categorizations of inflammatory myopathies that were previously simply known as polymyositis, dermatomyositis, and inclusion-body myositis.1 This review addresses the main pathological findings in various muscle diseases and provides the latest knowledge about muscle pathologies.
MUSCLE BIOPSY
A muscle biopsy should be performed when the results of full clinical and laboratory assessments support the presence of muscle diseases, such as progressive or sustained muscle weakness and atrophy, a family history of muscle weakness, high serum creatine kinase, and myopathic motor-unit potentials in needle electromyography. The muscles to be biopsied should be selected primarily based on the distribution of the muscle weakness. However, it is not recommended to biopsy severely affected muscles, since they are mostly replaced by fat and fibrotic tissues, nor slightly affected muscles, since their changes are likely to be too minor to be detected. Imaging methods such as computed tomography and magnetic resonance imaging have recently been used to evaluate the severity of muscle diseases. If the conditions are satisfied, the quadriceps, biceps, deltoid, or gastrocnemius can be biopsied. The biceps brachii is the most preferred because 1) there is a relatively small amount of subcutaneous tissue overlying the muscle, and 2) the arrangement of muscle fibers is consistently parallel and thus it is easy to inspect a cross section.
A muscle biopsy is usually performed under local anesthesia. After incising the overlying skin, a muscle specimen is acquired by dissecting the connective tissue surrounding the muscle. The muscle specimen should be fixed on a cork disc and then frozen as soon as possible in isopentane chilled with liquid nitrogen at -160°C.
NORMAL MUSCLE PATHOLOGY
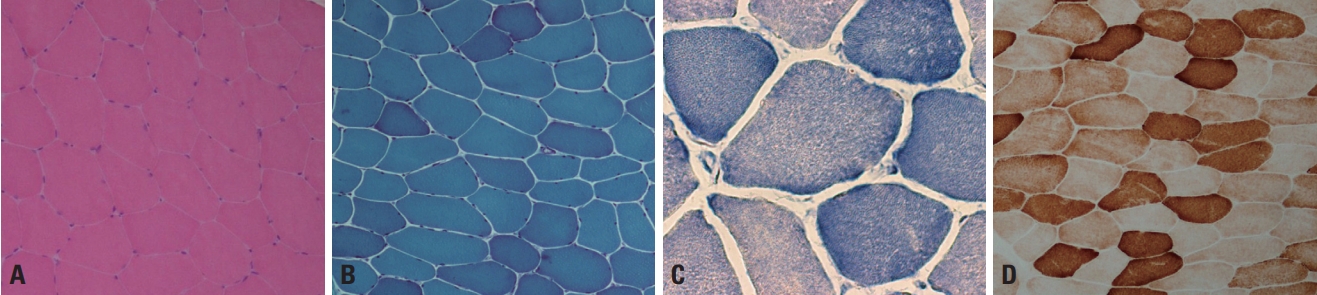
Normal muscle fibers are polygonal in shape and have sizes varying from 40 to 80 μm in adults and from 10 to 20 μm in infants.2 The fiber size increases with age, reaching the adult size by puberty.3 Muscle fibers are multinucleated cells formed by the fusion of postmitotic myoblasts, and multiple nuclei are located just beneath the sarcolemma.3 Each muscle fiber is enclosed by a small amount of connective tissue, called the endomysium. A group of muscle fibers forms a fascicle, which is separated from other fascicles by a perimysium. The perimysium contains peripheral nerves, muscle spindles, and small- or medium-sized vessels. These characteristics are well demonstrated by staining with hematoxylin and eosin stain or modified Gomori trichrome (mGT) stain (Fig. 1A, B). A muscle fiber includes regularly arranged myofibrils, with mitochondria and other cellular components present between the myofibrils. Oxidative enzyme stains such as nicotinamide dehydrogenase-tetrazolium reductase (NADH-TR), succinate dehydrogenase, and cytochrome c oxidase (COX) can be used to visualize the myofibrillar networks (Fig. 1C, D) and also reveal abnormalities of the mitochondria. In normal muscle fibers the sarcoplasm is homogeneously stained by oxidative enzymes, and a network arrangement of the myofibrils appears clearly in the high-power field (≥×400) of a light microscope (Fig. 1C).
Muscle fibers are categorized into types 1 and 2 according to their biochemical properties.2,3 Type 1 fibers have an oxidative metabolism, while a glycolytic metabolism is predominant in type 2. Type 2 fibers are further divided into type 2A, 2B, and 2C, where type 2A fibers exhibit partial oxidative activity and type 2C fibers are too immature to have a clear metabolic activity. In normal muscle pathology, the same types of fibers are randomly distributed in a checkerboard pattern rather than being grouped together (Fig. 2). The proportions of the different fiber types vary between muscles, with type 1, 2A, and 2B being well known to be present in approximately equal proportions in biceps brachii. Adenosine triphosphatase stain is useful for recognizing the distributions and proportions of the fiber types because each fiber type is differentially stained according to the acidity: type 2 fibers are darkly stained at pH 10.6 and type 1 fibers are darkly stained at pH 4.3 (Fig. 2A, B), while type 2A and 2B can be differentiated at intermediate pH values.
Acid phosphatase is present in macrophages and its staining is enhanced in inflammatory myopathies and muscular dystrophies but absent in normal muscles. Periodic acid-Schiff stain reveals the glycogen content within muscle fibers and is more intense in type-2 fibers having glycolytic metabolism (Fig. 2C). Oil red O stain reveals the neutral lipid content, and type-1 fibers are more stained more strongly since oxidative metabolism predominates (Fig. 2D). These stains are useful for detecting excessive glycogen and lipid droplets in glycogen and lipid storage diseases, respectively.
MUSCULAR DYSTROPHIES
Muscular dystrophies such as Duchenne muscular dystrophy (DMD) and Becker muscular dystrophy (BMD), and limb-girdle muscular dystrophies (LGMDs) are clinically characterized by progressive muscle weakness. Their onset ranges from early childhood to late adulthood according to the severity, with the exception of congenital muscular dystrophies (MDCs), which usually start immediately after birth or during infancy with motor developmental delay. The associated muscle weakness is predominantly in the proximal upper and lower limbs, and the facial muscles are usually spared. Serum creatine kinase is elevated in most types of muscular dystrophy, from twofold to up to several hundred times.4
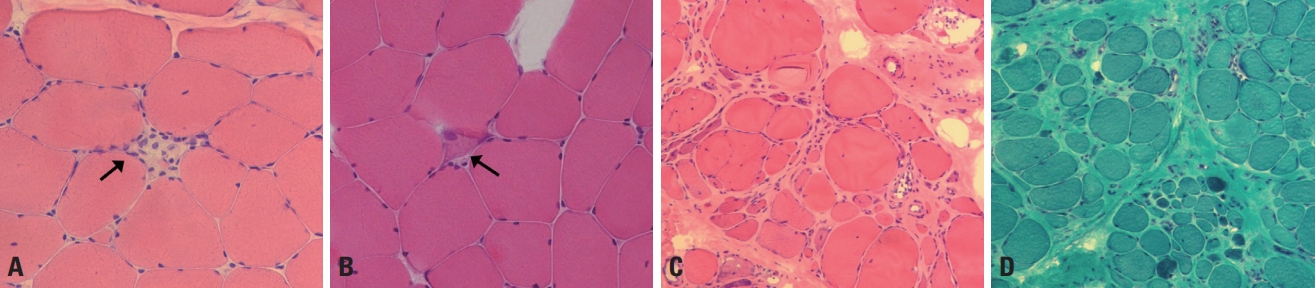
Muscular dystrophies are pathologically characterized by dystrophic changes. Dystrophic muscle pathology includes muscle fiber necrosis and regeneration, increased variations in fiber sizes, round fibers, and enhanced endomysial connective tissues (Fig. 3).4 These dystrophic changes are associated with cellular destruction. DMD/BMD and most LGMDs are caused by deficiency in or absence of the proteins that are responsible for the cellular architecture and structural stability of muscle fibers.5 Emery-Dreifuss muscular dystrophy and LGMD1B are caused by defects in nuclear proteins, such as emerin and lamin A/C, which increases the nuclear fragility and results in cellular destruction.6 Some muscular dystrophies such as MDC1A and Ullrich congenital muscular dystrophy are caused by defects in extracellular matrix proteins that ultimately give rise to instability of the muscle membranes.
Sarcolemma destruction leads to muscle fiber necrosis, which is, in turn, associated with the regeneration process. Necrotic muscle fibers have a pale cytoplasm and are named liquefied or hyaline fibers (Fig. 3A). Necrotic fibers are phagocytosed and surrounded by inflammatory cells. Regenerating muscle fibers have a basophilic cytoplasm and enlarged nuclei (Fig. 3B), which is due to protein synthesis being vigorous during regenerating myogenesis. Since the sequence of necrosis and regeneration repeatedly occurs as a chronic process in muscular dystrophies, the muscle fibers become rounded and connective tissues fill up the spaces between the muscle fibers. Over longer periods the atrophic and hypertrophic fibers become mixed, which markedly increases the variations in fiber sizes (Fig. 3C, D). These are common pathological findings in various types of muscular dystrophy. Accordingly, routine histological and histochemical stains are not helpful for differentiating the different types of muscular dystrophy.
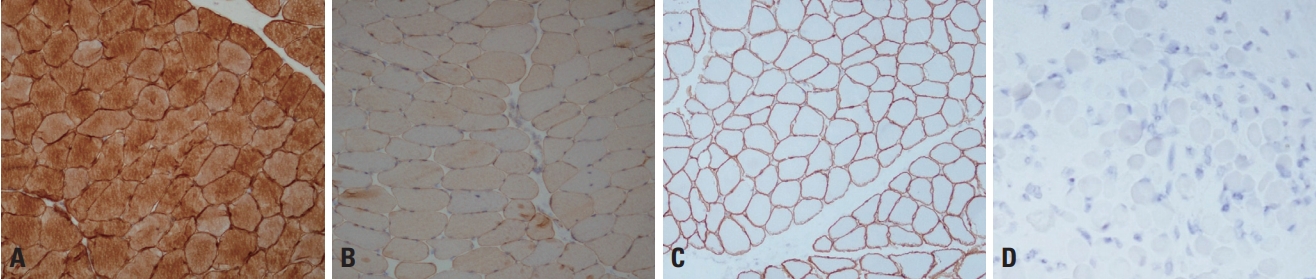
Immunohistochemistry (IHC) can be used to visualize the status of target proteins by the application of specific antibodies, and is useful for differential diagnoses of muscular dystrophies.5,7 In DMD caused by a total deficiency of dystrophin protein, IHC staining using the antibody against dystrophin is absent in sarcolemma. IHC can be used to examine the following target proteins: dysferlin in LGMD2B (Fig. 4A, B), sarcoglycans in LGMD2C-2F, α-dystroglycan in the muscular dystrophies caused by glycosylation defect of α-dystroglycan, and merosin in MDC1A (Fig. 4C, D). However, it should be noted that a secondary deficiency of another protein that strongly binds to the target protein by forming a complex can sometimes occur.7
CONGENITAL MYOPATHIES
Congenital myopathies are clinically defined as slowly progressive or nonprogressive muscle weakness after birth or during infancy, although a few cases do not start until adulthood. Weakness of facial and extraocular muscles are frequent manifestations, and early respiratory failure occurs in very severe cases. Motor developmental delay is common, and some patients never achieve the ability to walk. Serum creatine kinase is either slightly elevated or within the normal range.8
Muscle pathology can play an important role in the differential diagnosis of congenital myopathies. Each type of congenital myopathy has a specific pathological feature, as follows: nemaline bodies in nemaline myopathy, central cores in central core disease, and central nuclei in centronuclear myopathy and myotubular myopathy. In contrast to muscular dystrophies, congenital myopathies do not involve cellular destruction, and muscle fiber necrosis and regeneration rarely occur. Instead, the specific pathological features described above become significant diagnostic clues.8
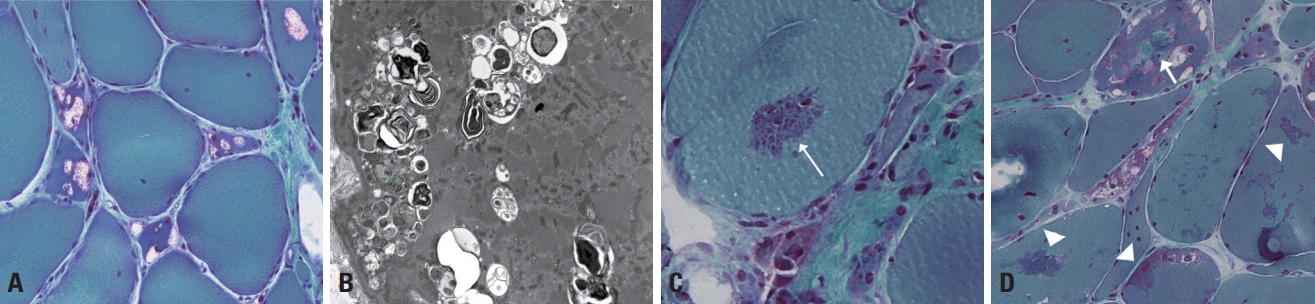
Nemaline bodies are a key feature of nemaline myopathy and are clearly revealed by mGT staining (Fig. 5A). These are red, have a dot-like shape, and are either clustered in subsarcolemmal regions or scattered throughout the sarcoplasm. In electron microscopy, nemaline bodies appear rod-shaped with an electron density similar to that of Z-disks, and the surrounding myofibrils are frequently disorganized (Fig. 5B). These findings are consistent with the causative genes of nemaline myopathy being associated with myofibrillar and Z-disk proteins, such as nebulin, actin, troponin, and tropomyosin. 9 Although nemaline myopathy is genetically heterogeneous, with nine known causative genes, its pathological presentation is generally not affected by the genotype.9,10 An exception occurs when TPM3 is the causative gene, for which a characteristic finding is nemaline bodies being present exclusively in type 1 fibers.11
Central cores refer to round, centrally located zones that exhibit deficiency in oxidative enzyme activities, which are clearly revealed by NADH-TR and COX staining (Fig. 5C). An ultrastructural finding is delineated regions where myofibrils are markedly disorganized (Fig. 5D). The presence of central cores in a majority of muscle fibers is suggestive of central core disease. However, physicians should take care given that nemaline bodies and central cores are not pathognomonic of nemaline myopathy and central core disease. Nemaline rods and cores are frequently found in myofibrillar myopathies, and core-rod myopathy is characterized by the simultaneous presentation of cores and rods.12 Sporadic late-onset nemaline myopathy can also occur, which is not a genetic disorder but can be associated with monoclonal gammopathy.13
The nuclei in centronuclear myopathy and myotubular myopathy are centralized or internalized within muscle fibers. Centronuclear myopathy may have an additional finding of radially arranged sarcoplasmic strands with nuclei as the center in NADH-TR staining (Fig. 5E). This is known to be genetically related to DNM2 being the causative gene.14,15 The term myotubular myopathy reflects that the muscle fibers with a single central nucleus mimic the myotubes during myogenesis. As a characteristic finding of myotubular myopathy, the presence of a peripheral halo indicates an oxidative enzyme-deficient region in the periphery of muscle fibers (Fig. 5F) that is known to be caused by the immaturity of myofibril formation. These findings suggest the usefulness of searching for the myotubularin 1 gene (MTM1).15
Along with their individual specific features, most congenital myopathies have the common finding that type 1 fibers are smaller and more prevalent. Meanwhile, congenital fiber type disproportion (CFTD) is characterized only by this manifestation, with no other structural abnormalities. To further satisfy the criteria of CFTD, type 1 fibers should be at least 12% smaller than type-2 fibers and constitute more than 50% of all the muscle fibers.8 Cap lesions, spheroid body, reducing body, and hyaline body can lead to diagnoses of cap myopathy, spheroid body myopathy, reducing body myopathy, and myosin storage disease, respectively, although all of these conditions are very rare.8
OTHER INHERITED MYOPATHIES
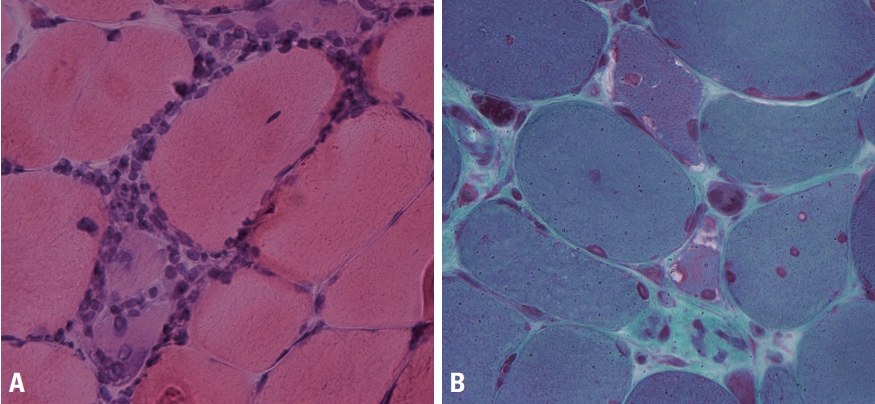
Vacuolar myopathy and myofibrillar myopathy are conditions in which the muscle pathology plays a significant role in their diagnoses. Vacuolar myopathy is characterized by autophagic vacuoles, which are clearly evident as rimmed vacuoles under a light microscope.16,17 Rimmed vacuoles appear as empty spaces rimmed by red granules in mGT staining (Fig. 6A), and their ultrastructural findings are typified by clusters of vacuoles containing amorphous, myeloid structures (Fig. 6B). Since the autophagic process involves the degradation of cellular organelles and other cytoplasmic contents, rimmed vacuoles can be found in any muscle diseases with excessive autophagic mechanisms, such as in Pompe disease (glycogen storage disease type II) and X-linked myopathy with excessive autophagy; however, these conditions are very rare. The most famous disease associated with rimmed vacuoles is GNE myopathy, which is caused by deficiency of the cytosolic protein GNE (a dual enzyme of UDP-N-acetylglucosamine epimerase/and N-acetylmannosamine kinase). The disease was named distal myopathy with rimmed vacuoles before the term GNE myopathy was decided based on the causative gene being GNE. Rimmed vacuoles are not pathognomonic, and can also be seen in LGMD1A, oculopharyngeal muscular dystrophy, and myofibrillar myopathies. Physicians need to consider detailed clinical information when differentiating various myopathies with rimmed vacuoles.
Myofibrillar myopathies are characterized by the presence of various types of cytoplasmic inclusions such as cytoplasmic, spheroid, and sarcoplasmic bodies, as well as rimmed vacuoles.16 Rimmed vacuoles occur frequently in myofibrillar myopathies since the autophagic process is augmented by massive degradation of disrupted myofibrillar components. mGT stain is the most useful for demonstrating cytoplasmic inclusions, with which they appear either round (cytoplasmic and spheroid bodies) or irregular in shape (sarcoplasmic body), and either deep red (cytoplasmic body) or green (spheroid and sarcoplasmic bodies) (Fig. 6C, D).16 Electron microscopy reveals the massive disruption of myofibrillar structures, Z-line streaming, and Z-disk accumulation. The diseases categorized as myofibrillar myopathy involve defects in sarcomeric proteins, such as desmin, αB crystalline, filamin C, Z-line alternatively spliced PDZ-motif containing protein (ZASP), myotilin, four-and-a-half-LIM domain 1 protein (FHL1), and BCL2-associated athanogene 3 (BAG3). IHC can be used to assess the accumulation of these proteins.
IDIOPATHIC INFLAMMATORY MYOPATHIES
The old classification of idiopathic inflammatory myopathy was based on the criteria of Bohan and Peter developed in 1975, which classified idiopathic inflammatory myopathies into polymyositis and dermatomyositis.18 Griggs et al.19 added inclusion-body myositis to the existing classification in 1995. There have been considerable limitations in applying these old criteria in diagnostics and therapeutics. The improved pathological understanding and identification of myositis-specific antibodies has led to a new classification of idiopathic inflammatory myopathy being proposed.1,20
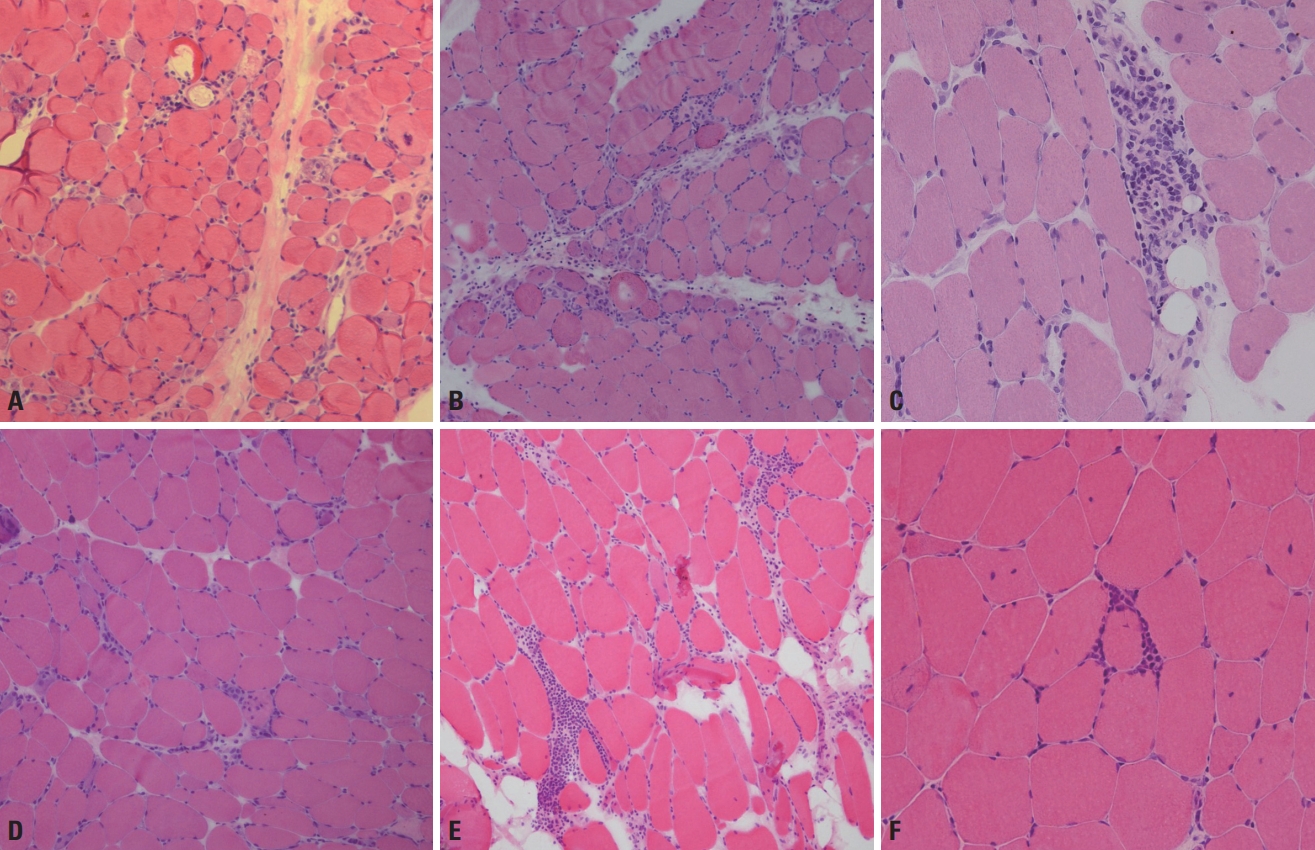
Dermatomyositis is pathologically characterized by perifascicular atrophy (Fig. 7A), which was also included in the criteria of Bohan and Peter but in a less-specific manner.20 Moreover, deposition of the membrane attack complex around capillaries and vacuolated fibers are evident in dermatomyositis. In antisynthetase syndrome, perifascicular necrosis (Fig. 7B) is more remarkable than perifascicular atrophy.21 This syndrome is actually a serological disease entity defined by the presence of antibodies against aminoacyl transfer RNA synthetases, such as anti-Jo-1, anti-PL-7, and anti-PL-12 antibodies. Dermatomyositis and antisynthetase syndrome share the pathological features of perifascicular atrophy and perimysial and perivascular pathologies (Fig. 7C).
Immune-mediated necrotizing myopathy shows frequent necrotic and regenerating fibers and scattered macrophage infiltration (Fig. 7D). There is no definite focus of inflammatory cellular infiltration either in the endomysium or perimysium, which result in it frequently being misdiagnosed as muscular dystrophy. The old criteria classified these cases as polymyositis, despite inflammation being sparse. Based on the new classification, authentic polymyositis seems very rare. Polymyositis is pathologically characterized by the presence of endomysial inflammatory infiltration (Fig. 7E) and inflammatory cells surrounding or invading nonnecrotic fibers (Fig. 7F), as well as necrotic and regenerating processes.
In addition to necrotic/regenerating fibers and endomysial inflammation (Fig. 8A) the presence of rimmed vacuoles (Fig. 8B) is a major pathological feature in inclusion-body myositis, although it is not always present. Cases without rimmed vacuoles have also been classified into polymyositis based on the old criteria. COX-negative fibers and ragged red fibers suggestive of mitochondrial abnormalities can be found in inclusion body myositis.
NEUROGENIC CHANGES
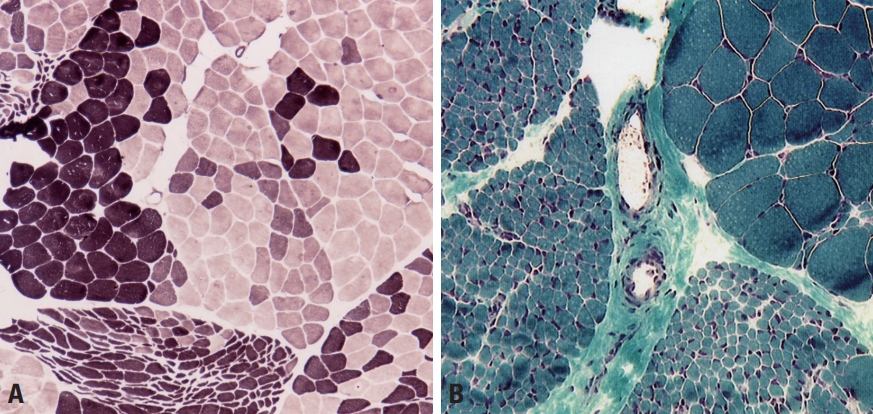
Neurogenic muscle weakness can manifest as sensory deficit, a distal-dominant distribution of muscle weakness, and findings of electrophysiological studies before deciding about whether to perform a muscle biopsy. However, differentiating between myopathic and neurogenic weakness is not always easy. We frequently see neurogenic changes in muscle pathology, and the most remarkable finding is fiber type grouping where the same types of muscle fibers become grouped through denervation and reinnervation processes in neurogenic disorders (Fig. 9A). Examples include motor neuron diseases such as spinal muscular atrophy and amyotrophic lateral sclerosis, as well as chronic or inherited polyneuropathies such as Charcot-Marie-Tooth disease. The increasingly active denervation process results in grouped muscle fibers being denervated again, resulting in grouped atrophy or fascicular atrophy (Fig. 9B). In neurogenic changes, type-2 fibers are more atrophied and atrophic fibers become angulated.
CONCLUSIONS
Muscle pathology findings can provide diagnostic clues and also help to understand the pathological processes causing muscle weakness. Inspections of muscle pathology can reveal whether the necrotic process is progressing, whether the inflammatory process contributes to a pathological process, if it is acute or chronic, and if there are neurogenic or myopathic changes. However, the findings for muscle pathology are not always definitive, and so care is necessary with interpreting these findings, with it being useful to consider them in combination with the clinical manifestations.